




TBP/TATA Binding Protein (4H2) Mouse mAb,HRP)
-YM2149

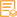


主要信息
Target
TBP/TATA Binding Protein
Host Species
Mouse
Reactivity
Human, Rat, Mouse
Applications
WB
MW
38kD (Calculated)
Conjugate/Modification
HRP



详细信息
推荐稀释比
Optimal working dilutions should be determined experimentally by the investigator; Suggested starting dilutions are as follows:WB 1:1000-2000.
组成
Liquid in PBS, pH 7.4, containing 0.02% sodium azide as preservative and 50% Glycerol.
特异性
TBP/TATA Binding Protein Monoclonal Antibody(4H2) HRP Conjugated, specially designed for your Western blot analysis.
纯化工艺
The antibody was affinity-purified from mouse ascites by affinity-chromatography using specific immunogen.
储存
Stable for one year at -15°C to -25°C from date of shipment. For maximum recovery of product, centrifuge the original vial after thawing and prior to removing the cap. Aliquot to avoid repeated freezing and thawing.
浓度
1mg/ml
理论分子量
38kD
偶联
HRP
克隆性
Monoclonal
克隆号
4H2
同种型
IgG
相关产品
Primary Antibodies
TFIID Rabbit pAb
YT4615
预览→
Primary Antibodies
TBP/TATA Binding Protein (1F6) Mouse mAb
YM3515
预览→
Primary Antibodies
TBP/TATA Binding Protein (4H2) Mouse mAb
YM3511
预览→
Primary Antibodies

TBP/TATA Binding Protein (4H2) Mouse mAb,HRP)
YM2149
预览→
抗原&靶点信息
特异性:
TBP/TATA Binding Protein Monoclonal Antibody(4H2) HRP Conjugated, specially designed for your Western blot analysis.
展开内容
基因名称:
TBP
展开内容
蛋白名称:
TATA-box-binding protein (TATA sequence-binding protein) (TATA-binding factor) (TATA-box factor) (Transcription initiation factor TFIID TBP subunit)
展开内容
别名:
TBP
展开内容
背景:
Initiation of transcription by RNA polymerase II requires the activities of more than 70 polypeptides. The protein that coordinates these activities is transcription factor IID (TFIID), which binds to the core promoter to position the polymerase properly, serves as the scaffold for assembly of the remainder of the transcription complex, and acts as a channel for regulatory signals. TFIID is composed of the TATA-binding protein (TBP) and a group of evolutionarily conserved proteins known as TBP-associated factors or TAFs. TAFs may participate in basal transcription, serve as coactivators, function in promoter recognition or modify general transcription factors (GTFs) to facilitate complex assembly and transcription initiation. This gene encodes TBP, the TATA-binding protein. A distinctive feature of TBP is a long string of glutamines in the N-terminus. This region of the protein modulates the DNA bin
展开内容
功能:
Disease:Defects in TBP are the cause of spinocerebellar ataxia type 17 (SCA17) [MIM:607136]. Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA17 is an autosomal dominant cerebellar ataxia (ADCA) characterized by widespread cerebral and cerebellar atrophy, dementia and extrapyramidal signs. The molecular defect in SCA17 is the expansion of a CAG repeat in the coding region of TBP. Longer expansions result in earlier onset and more severe clinical manifestations of the disease.,Function:General transcription factor that functions at the core of the DNA-binding multiprotein factor TFIID. Binding of TFIID to the TATA box is the initial transcriptional step of the pre-initiation complex (PIC), playing a role in the activation of eukaryotic genes transcribed by RNA polymerase II.,polymorphism:The poly-Gln region of TBP is highly polymorphic (25 to 42 repeats) in normal individuals and is expanded to about 47-63 repeats in spinocerebellar ataxia 17 (SCA17) patients.,similarity:Belongs to the TBP family.,subunit:Belongs to the TFIID complex together with the TBP-associated factors (TAFs). Component of the transcription factor SL1/TIFIB complex, composed of TBP and at least TAF1A, TAF1B TAF1C, and TAF3. Binds DNA as monomer. Interacts with TAFs, TFIIB, NCOA6, DRAP1, DR1 and ELF3. Interacts with SPIB, SNAPC1, SNAPC2 and SNAPC4. Interacts with HIV-1 Tat. Interacts with UTF1 which acts as a coactivator of ATF2 transcriptional activity. Interacts with GPBP1 (By similarity). Interacts with BRF2.,tissue specificity:Widely expressed, with levels highest in the testis and ovary.,
展开内容
细胞定位:
Nucleus .
展开内容
组织表达:
Widely expressed, with levels highest in the testis and ovary.
展开内容
研究领域:
>>Basal transcription factors ;
>>Huntington disease ;
>>Spinocerebellar ataxia ;
>>Human papillomavirus infection ;
>>Human T-cell leukemia virus 1 infection ;
>>Viral carcinogenesis
>>Huntington disease ;
>>Spinocerebellar ataxia ;
>>Human papillomavirus infection ;
>>Human T-cell leukemia virus 1 infection ;
>>Viral carcinogenesis
展开内容
信号通路
文献引用({{totalcount}})
Recently Viewed Products
Clear all







Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}